Welcome to European Federation of Societies for Ultrasound in Medicine and Biology
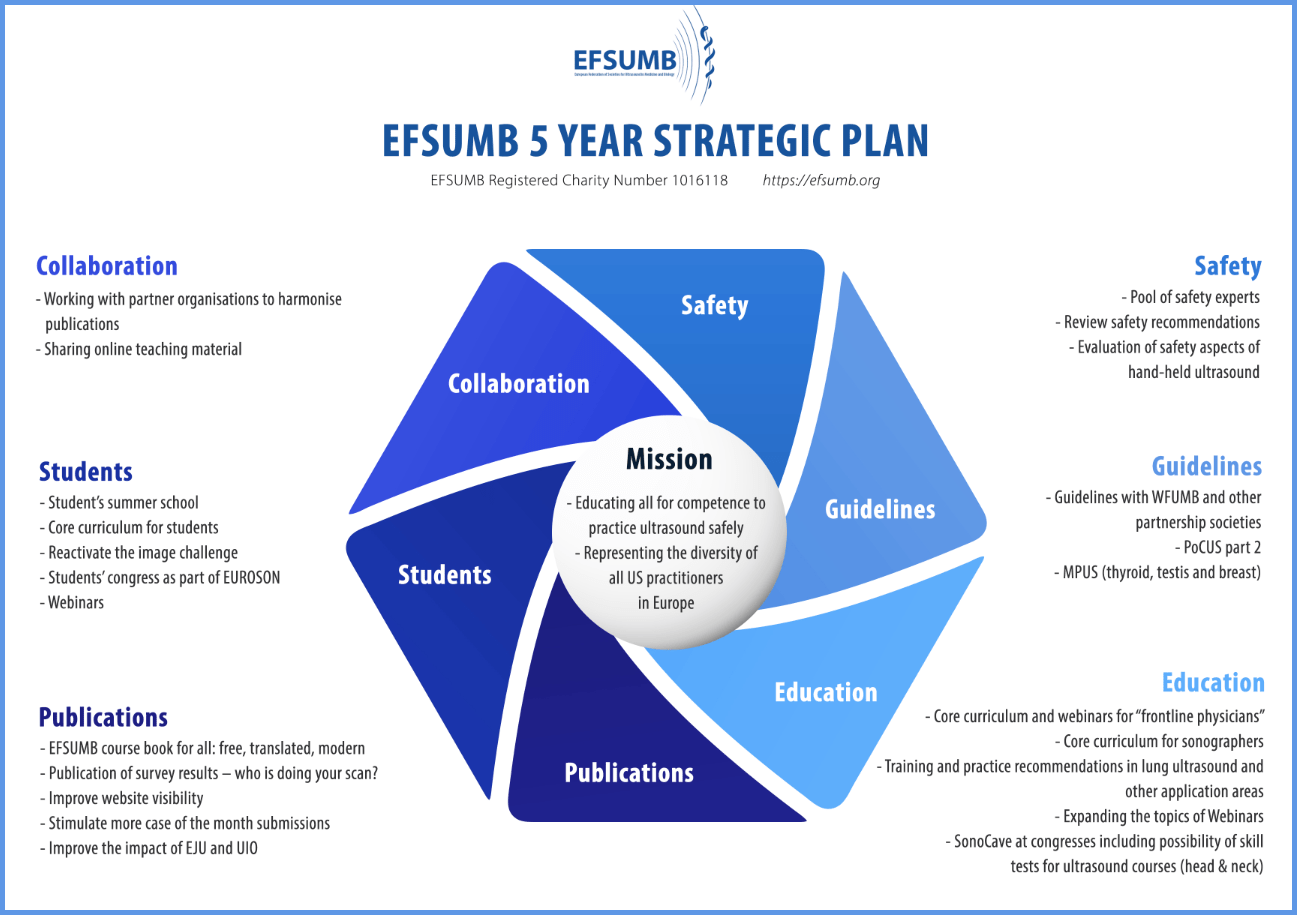
Educating all for competence to practice ultrasound safely
European Federation of Societies for Ultrasound in Medicine and Biology (EFSUMB): Call for peace
The Russian war of aggression against Ukraine has now been raging for over 600 days. Hundreds of thousands of people have already died, not only soldiers, but also women and children, patients in hospitals, mothers with their newborns and old people in nursing homes. Now, an unimaginably brutal terrorist attack by Hamas has cost the lives of so many civilian citizens and visitors to Israel in one day, shattered hopes as not since the Shoah, taken hostages, not only those taken from the territory of Israel, but also its own people, the Palestinian families living in the Gaza Strip. Hundreds of thousands resettled or forced to flee, countless human tragedies, lives senselessly destroyed. All these human lives have the same value, are irreplaceable, independently of age, gender, religion, nation or political belief. Read our full statement here >







22Apr
02May
EUROSON SCHOOL: Basic Ultrasound Course
Antalya, Turkey
14May
EFSUMB Webinar: Vascular
Online Virtual Event
18Jun
Chinese Webinar Series: Transcavity CEUS
Online Virtual Event
No event found!


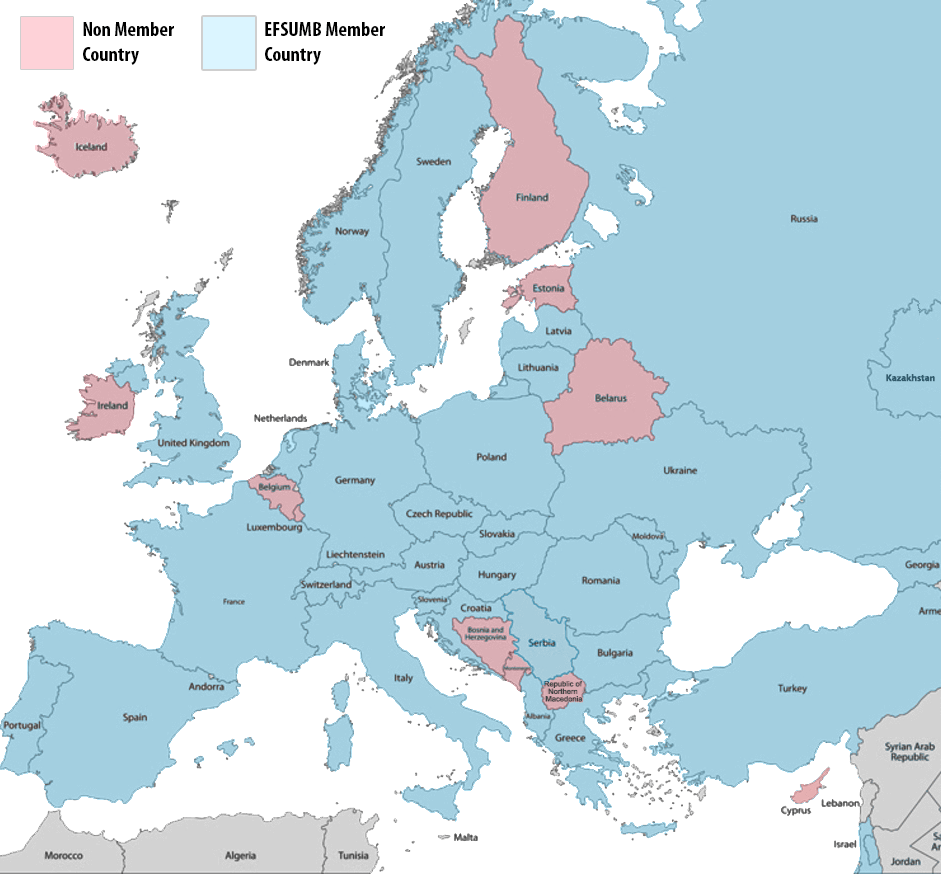
Whether you are based in Europe or not, EFSUMB is now inviting members from across the world. Join a network of 31 countries and hundreds of individual members to support the continuous education for competence to practice ultrasound safely.
If you are a member of a National Society located in Europe, it is likely you are already a member of EFSUMB. Check your status using one of the links below.
Benefits of EFSUMB Membership
Individual Membership
Student Membership Affiliate Society Membership
Already an EFSUMB Member? Members Log In Here